
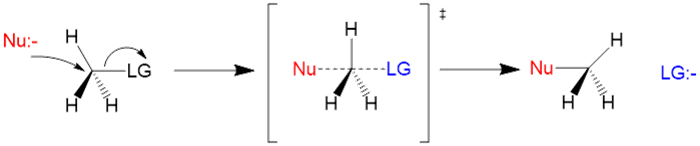
We learn the two mechanisms of nucleophilic substitution in school, namely SN1 and SN2. To recap, SN1 is a two-step mechanism involving first the leaving of the leaving group to form a carbocation intermediate, which is then attacked by the nucleophile giving a racemic mixture of products. SN2 is a one-step mechanism involving the simultaneous leaving of the leaving group and attack of the nucleophile, resulting in inversion of the carbon centre.
SN1 mechanism
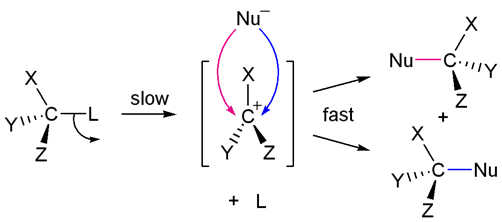
SN2 mechanism
We also learn that more alkyl substituents on the electrophilic carbon favours the SN1 mechanism, as the alkyl substituents stabilise the carbocation and the increased sterics disfavours SN2. In this tutorial, we will discuss more factors which affect whether the mechanism goes by the SN1 or SN2 pathway.
Firstly, the strength of the attacking nucleophile affects the mechanism of nucleophilic substitution. As the nucleophile features in the rate determining step of an SN2 reaction, a strong nucleophile favours SN2, while a weak nucleophile favours SN1 by decreasing the rate of the SN2 pathway. Generally, the more available the lone pair, the stronger the nucleophile. We will delve deeper into the factors that affect nucleophile strength soon.
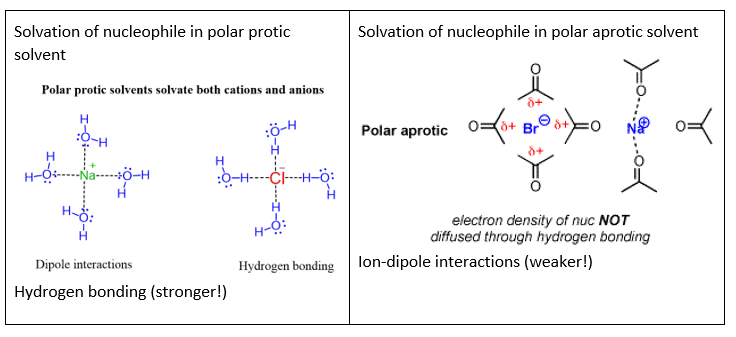
Secondly, the solvent used also affects the mechanism of nucleophilic substitution. As stated above, a strong nucleophile (which is usually an anionic species) favours SN2. The nature of the solvent affects the strength of the nucleophile in the reaction mixture. Polar protic solvents which are capable of hydrogen bonding interact strongly with the anionic nucleophile, surrounding it with a shell (or a “cage”) of solvent molecules. This solvation shell reduces the strength of the nucleophile, causing the SN1 reaction to be favoured. By contrast, polar aprotic solvents which lack the ability to form hydrogen bonds interact less strongly with the anionic nucleophile, and since the nucleophile is less tightly solvated, it is stronger and SN2 is favoured.
The following table shows a summary of the reaction conditions which favour SN1 and SN2 pathways:
| SN1 | SN2 |
| More substituted/sterically hindered carbon | Less substituted/sterically hindered carbon |
| Weaker nucleophile | Stronger nucleophile |
| Polar protic solvents | Polar aprotic solvents |
Neighbouring group participation in substitution
For the substitution of a chiral halogenoalkane, if the reaction produces mostly the inverted product, then we can say that SN2 predominates. If the reaction produces both enantiomers in roughly equal proportions, then we can say that SN1 predominates. But what happens if we observe that the reaction produces mostly the uninverted product? Is that even possible?
The answer is yes, if neighbouring group participation results in two successive SN2 substitutions leading to the formation of the eventual product. An example is shown below, where the substitution of I by Br goes through two SN2 reactions due to the participation of a neighbouring sulfur atom:
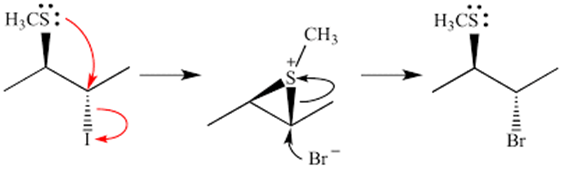
As inversion about the chiral centre occurs twice, the eventual product mixture will contain mostly the uninverted product.
Thus far, we have kept in line with the H2 conclusion that in an SN1 reaction, the two enantiomers will be produced in equimolar amounts. However, we do observe that this is not always the case, due to the ion-pair effect. When the leaving group leaves as a negatively charged group, it could remain in the vicinity of the positively charged carbocation, thus shielding one side from nucleophilic attack, resulting in a greater formation of the inverted product, as shown below:
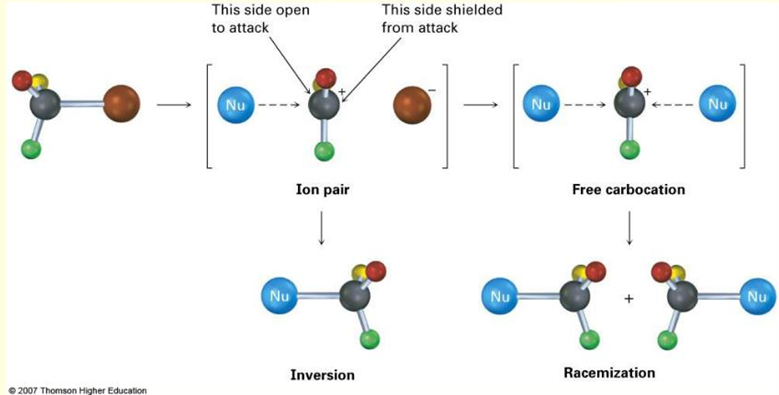
Hence in reality, SN1 reaction usually forms a slightly greater proportion of the inverted product.
SN2’ substitution of allylic halogen groups
We learn in the H2 syllabus that the substitution of allylic halides proceed by SN1 mechanism, since the allylic carbocation is stabilised by resonance. However, did you know of a different mechanism that results in the formation of another side-product? This mechanism is called SN2’, also known as nucleophilic conjugate substitution, where interestingly, the nucleophile attacks the C=C double bond instead of the carbon bonded to the halogen atom:

The reason for this interesting site of attack is because the presence of the nearby electronegative -Cl group lowers the energy of the pi antibonding MO of the C=C bond, thus allowing it to be attacked. Note that the alkene carbon being attacked, however, is a soft electrophile relative to the carbon bonded to chlorine (as it has a smaller partial positive charge), hence SN2’ is favoured by soft nucleophiles (such as the large and polarisable S atom, in this case).
Elimination: E1 and E2
In the school syllabus, we also learn that halogenoalkanes can undergo elimination reactions, producing alkenes by the elimination of HX. Now, we will take a look at the mechanism of such elimination reactions. In the E1 mechanism, the halide group leaves as X–, forming a carbocation intermediate. A base then deprotonates an adjacent carbon, resulting in the formation of an alkene. As the rate determining step is the first step involving only the halogenoalkane, rate=k[halogenoalkane].

In the E2 mechanism, the leaving of the halide and the deprotonation of the adjacent carbon by a base occurs in one concerted step. As the reaction is now an elementary reaction involving both the base and the halogenoalkane, the reaction is overall second order with rate=k[base][halogenoalkane].
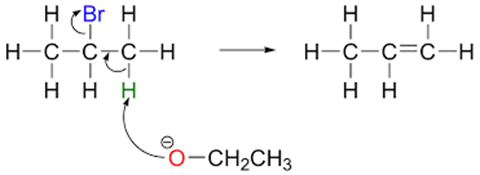
An interesting stereochemical characteristic of the E2 reaction is that it requires an antiperiplanar arrangement of the H and X atoms to occur. That is to say, the H and X atoms must be along the same plane, but anti to each other.
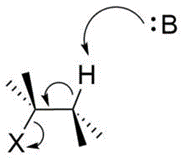
This is so that the bonding MO of the C-H bond can overlap with the antibonding MO of the C-X bond, resulting in the formation of a pi bond and the breaking of the C-X bond.
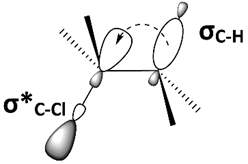
Therefore, E2 is stereospecific as the requirement for antiperiplanar elimination means only one stereoisomer of the alkene is produced.
When an antiperiplanar arrangement is not possible, say due to restricted rotation about a ring structure, then E2 reaction is unlikely, but not impossible if there is a synperiplanar arrangement.
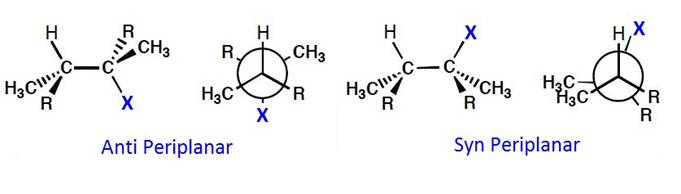
However, the transition state for synperiplanar E2 elimination has an eclipsed geometry, leading to it being higher in energy and thus a greater activation energy of the reaction.
Similar to SN1 versus SN2 mechanisms, the reactants involved and the reaction conditions determine whether E1 or E2 is the favoured mechanism for an elimination reaction. As E1 involves the formation of a carbocation intermediate, the ability to form a stable carbocation intermediate (tertiary or stabilised by resonance) will favour E1 elimination.
As the base appears in the rate equation for E2 elimination, the stronger the base, the more E2 is favoured over E1. And since solvation by a polar protic solvent reduces the strength of a base, a polar protic solvent favours E1 elimination, while a polar aprotic solvent favours E2 elimination.
While in SN2, the presence of alkyl substituents hinders substitution by means of steric repulsion, E2 reactions (and E1 reactions as well) are actually favoured by the presence of more alkyl groups, as it stabilises the resultant alkene product by Zaitsev’s Rule. Thus generally the more substituted alkene will be produced in greater quantities in elimination reactions.
Competition between substitution and elimination
As mentioned above, substitution of halogenoalkanes occurs by attack of a nucleophile, while elimination of halogenoalkanes occurs by attack of a base. But can a nucleophile act as a base, and vice versa? The answer is yes, since both involve donation of a lone pair to an electron deficient species (a δ+ C in the case of a nucleophile, and a H+ in the case of a base). Hence there is always inherent competition between nucleophilic substitution and elimination of halogenoalkanes. In fact, SN1 and E1 share the same carbocation intermediate!
But is a stronger nucleophile always a stronger base? One might be tempted to say yes, given the close parallel between a nucleophilic attack on C and a base abstracting an acidic proton. Indeed we do see that the presence of a negative charge enhances both nucleophilicity and basicity, as it increases the electron density on the atom donating the lone pair. We also see that when comparing atoms of similar size, the more electropositive atom is both more nucleophilic and also more basic, as the lone pair(s) are more available for donation.
However, when it comes to the effect of the size of the atom and sterics, we actually see opposite trends in nucleophilic and basic strength. As the size of the atom increases, nucleophilicity generally increases, as the orbitals of the nucleophile can better overlap with the relevant LUMO of the halogenoalkane. This is because a larger atoms have a HOMO of higher energy (since the electrons are less strongly attracted to the nucleus), allowing it to overlap well with the LUMO of the halogenoalkane, which is usually the sigma antibonding MO of the C-X bond. In addition, for negatively charged nucleophiles in a polar protic solvent, larger nucleophiles are less tightly solvated, and thus act as stronger nucleophiles. However, when the size of the atom increases, basicity generally decreases, as the negative charge density is reduced, meaning that the base attracts an acidic proton less strongly. We can see this trend at play in the halides, where going down the group from F– to I–, basicity decreases but nucleophilicity increases.
Lastly, nucleophilicity is greatly reduced by steric hindrance caused by a bulky nucleophile, as the nucleophilic atom needs to approach close enough to the δ+ C of the halogenoalkane (itself already hindered by its substituents) in order to attack. In contrast, basicity is not much affected by steric hindrance, since acidic protons are not as sterically hindered and can be abstracted readily even by a bulky base. The table below summarises the above:
| Nucleophilicity in SN reactions | Basicity | |
| Definition | Affinity of the lone pair for a carbon atom. | Affinity of the lone pair for a proton (H+) |
| Effect of electric charge | Negatively charged atoms are better nucleophiles | Negatively charged atoms are better bases |
| Effect of electronegativity | More electronegative atoms are worse nucleophiles (when size is similar) | More electronegative atoms are worse bases (when size is similar) |
| Effect of size of atom | Larger atoms are better nucleophiles | Larger atoms are worse bases |
| Effect of sterics | Steric hindrance decreases nucleophilicity | Sterics have little effect on basicity |
Now, we are well-prepared to answer the question bugging us: if there is constantly competition between substitution and elimination, how do we favour one over the other? If we want to favour elimination, the key is to use a good base which is a poor nucleophile. Usually, a bulky base does the trick, as the steric hindrance provided by large substituents hinders its ability to act as a nucleophile, without interfering with its basic strength. Examples of bulky bases are shown below:
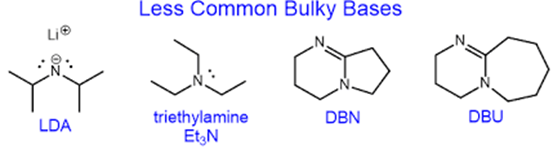
We can now answer the reason for the different conditions for elimination (NaOH in ethanol) and nucleophilic substitution by OH– (NaOH (aq)) we learn in school. The use of a polar protic solvent like water actually decreases basic strength to a greater extent than it decreases nucleophilicity, hence aqueous OH– act as a poor base but decently good nucleophile, allowing for the aqueous hydroxide to substitute a halogenoalkane, even though unsolvated OH– is actually a very strong base.






