
What is MO Theory?
Beginner learners of more advanced organic chemistry would certainly have encountered molecular orbital theory in some form, and may even have developed a certain aversion or fear of this powerful theory. No doubt, the intricacies of the theory lies in its quantum mechanical and mathematical foundation, but I will just go into a qualitative picture of the theory.
As you might know, atoms contain atomic orbitals (AOs), which hold electrons. These AOs can be described by individual orbital wavefunctions. In a molecule, atoms come together, and their atomic orbitals combine quantum mechanically to form molecular orbitals (MOs). According to the linear combination of atomic orbitals (LCAO) theory, the orbitals can combine either constructively or destructively, forming a bonding MO and an antibonding MO respectively. The bonding MO has electron density in the internuclear region, and occupation of the bonding MO is stabilising, and indicates formation of a bond. In contrast, the antibonding MO has electron density pointing away from the internuclear region, and occupation of the antibonding MO is destabilising.
The bond order of a molecule can be calculated by the following equation:
Bond order =0.5*(number of electrons in bonding MOs – number of electrons in antibonding MOs)
In diatomic hydrogen, the two hydrogen 1s AOs combine to form a bonding and antibonding MO. According to LCAO, n number of AOs combine to form n number of MOs. As these MOs are formed from the direct overlap of the 1s AOs, they are labelled as sigma (σ) MOs, with the * indicating an antibonding MO. We fill the lower energy MO with the two electrons of the molecule. The bond order of diatomic hydrogen, applying the above formula, will then be 0.5*2=1, confirming what we expect for the H2 molecule.

Molecular orbital diagram for diatomic hydrogen
(Source: http://www.sparknotes.com/chemistry/bonding/molecularorbital/section1.rhtml)
For contrast, let us take a look at diatomic helium. In diatomic helium, the two 1s AOs of He combine to form a bonding σ MO and an antibonding σ* MO. We allocate the four electrons of the molecule, filling up both MOs.
The bond order of diatomic helium, applying the formula once again, would then be 0.5*(2-2)=0, confirming what we would expect, that He does not form He2 molecules.
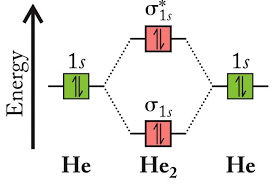
Molecular orbital theory for diatomic helium.
(Source: https://people.wou.edu/~postonp/ch221/pdf/Summary-MO-Theory-w16.pdf)
Ultimately, MO theory is a quantum-mechanical theory, built upon mathematical principles. The LCAO process is also a mathematical one, but can be qualitatively understood as constructing the resultant MOs from a weighted sum of the individual AOs interacting together. Consider the case of two AOs, ψA and ψB forming a bonding and an antibonding MO. The LCAO process to form the bonding MO can be represented by the following equation:
Bonding MO=a*ψA + b*ψB where a + b = 1
As we would want to minimise the energy of the bonding MO as much as possible, if the energy of ψA is lower than ψB , then ψA would contribute more to the bonding MO, and the coefficient a will be greater than b. The greatest stabilisation occurs when the two AOs are similar in energy, hence ideal MO interactions arise when the interacting orbitals are similar in energy.
Frontier MO Theory
For large molecules, given the large number of AOs interacting, there would be many, many MOs. Thus, a complete and rigorous MO theory analysis would no longer be convenient to use in a practical sense. The solution to this is to just consider the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). These orbitals are termed the frontier orbitals of the molecule. In fact, most reactions can be well predicted with considerations of the interaction between these frontier orbitals, keeping in mind that two MOs need to have similar energy and compatible geometry to have effective interactions.
HOMO, LUMO, and reactivity
Why can Br2 (and other halogens) act as an electrophile? Br2 has an unoccupied energetically low-lying sigma antibonding MO, due to the electronegativity of bromine, thus allowing it to use that low-energy unoccupied MO to accept a lone pair of electrons and hence act as an electrophile. Another reason for the low-lying sigma antibonding orbital would be due to the diffuse atomic orbitals of Br2 used to form the sigma bonding molecular orbital; the poor overlap results in the sigma antibonding orbital not being raised in energy that much.
Why can an alkene act as a good nucleophile? The occupied C-C pi bonding MO is relatively high in energy (due to less effective orbital overlap in pi bonding compared to sigma bonding), hence allowing the electrons residing in that high-energy occupied MO to be donated to an electrophile.
Hence we see how HOMO and LUMO energy levels affect reactivity. As Claydens’ Organic Chemistry succinctly puts it, “The best nucleophiles have high-energy occupied molecular orbitals (HOMOs). The best electrophiles have low-energy unoccupied molecular orbitals (LUMOs).”
The stability of molecules with long carbon chains is due to the unreactivity of the C-C bonds. Why do most of the “exciting chemistry” occur at bonds with heteroatoms? This is because C-C bonds have low energy occupied MOs and high energy unoccupied MOs, making them unreactive. A carbon bonded to an electropositive atom allows for a high-energy occupied MO, while a carbon bonded to an electronegative atom allows for a low-energy unoccupied MO, making it accessible for electrophiles and nucleophiles respectively.
Example: Nucleophilic addition at the carbonyl group
This is a most classic reaction involving compounds containing the carbonyl group. In most introductory organic chemistry texts on this reaction, it is just said that a nucleophile attacks the electron deficient C atom, causing the cleavage of the carbon-oxygen pi bond. However, a more wholesome understanding can be achieved when one takes into account the frontier molecular orbital interactions occurring in this reaction.
There are few organic molecules that has energetically low-lying atomic orbitals that are unoccupied (empty AOs), but instead the lowest unoccupied molecular orbitals (LUMOs) are usually the antibonding orbitals that are associated with electronegative atoms, as these electronegative atoms help to lower the energy of the molecular orbitals. Thus in this case, the ability of the carbonyl carbon in acting as an electrophile is explained using the electronegativity of the oxygen atom lowering the energy level of the pi antibonding orbital (the LUMO). This makes sense because the partial positive charge on the carbonyl carbon explains why negatively charged nucleophiles will be drawn close to facilitate the reaction, but does not explain why the carbonyl carbon is able to accept donated electron density (despite having a complete valence shell).
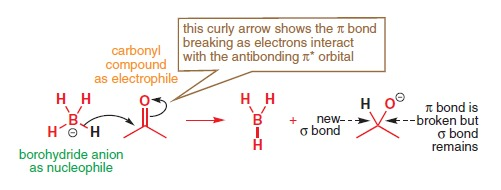
Source: Clayden’s Organic Chemistry
Such a perspective that is enriched by FMO theory also yields interesting insights. As the pi antibonding orbital accepts electron density, the carbon-oxygen pi bond is destabilised and cleaves. This allows for the formation of an alcohol group following the protonation of the alkoxide ion.
FMO theory can also shed insight on why the nucleophile attacks the carbonyl carbon and not the oxygen atom. Since the oxygen AO has lower energy than the carbon AO due to the greater electronegativity of oxygen, the pi bond can be described by a linear combination of the oxygen and carbon AOs with the greater coefficient on oxygen. In converse, the pi antibonding orbital has the greater coefficient on the carbon atom, hence a more effective overlap between the HOMO of the nucleophile and the pi antibonding orbital would be achieved when the nucleophile approaches the carbonyl group on the side of the carbonyl carbon.
Hence we see the benefits of considering frontier MO theory in analyzing organic chemistry reaction, in this case being nucleophilic addition.






